
On February 2, 2026, 21 CFR 820.30 became a reserved section. Every obligation governing ISO 13485 design controls for FDA-regulated medical devices now sources from ISO 13485:2016 Clause 7.3, incorporated by reference under 21 CFR 820.10(a). Manufacturers who relabeled DHF section headers to match ISO clause numbers and stopped there hold structurally non-conforming records — records that satisfied legacy 820.30 but miss substantive requirements at Clauses 7.3.3, 7.3.6, and 7.3.7.
The gaps are not cosmetic. They are evidentiary.
What Changed: 820.30 Is Gone, Clause 7.3 Is the Regulation
The QMSR final rule did not simply “align” FDA requirements with ISO 13485. It replaced the regulatory text. Design control requirements under 21 CFR Part 820 are now ISO 13485:2016 Clause 7.3.1 through 7.3.10, made enforceable through 21 CFR 820.10(a). The incorporated documents — ISO 13485:2016 and ISO 9000:2015(E) Clause 3 — are identified at 21 CFR 820.7.
The structural mapping from legacy 820.30 to Clause 7.3 is roughly one-to-one: 820.30(a) maps to 7.3.1, 820.30(b) to 7.3.2, and so on through 820.30(j) to 7.3.10. But “roughly” is where organisations get caught. Three sub-clauses carry requirements that did not exist in their legacy counterparts, and a fourth has a crosswalk anomaly that no FDA guidance document has formally addressed.
FDA investigators under QMSR inspect directly against ISO 13485:2016 clause requirements. They do not accept ISO 13485 certification certificates or third-party CB audit reports as evidence of compliance.
The inspection still happens against the clause text — it just happens to be ISO clause text now.
Where Legacy DHF Records Break: Three ISO 13485 Design Control Gaps
Clause 7.3.3: Design Inputs Expanded Beyond 820.30(c)
Legacy 820.30(c) required design inputs to establish physical and performance requirements. That was the scope. Clause 7.3.3 carries the same baseline but adds three mandatory input categories:
Usability requirements according to intended use. Legacy design input records that defined functional specifications without explicit usability engineering references — IEC 62366-1 requirements, for example — do not satisfy Clause 7.3.3 on this point.
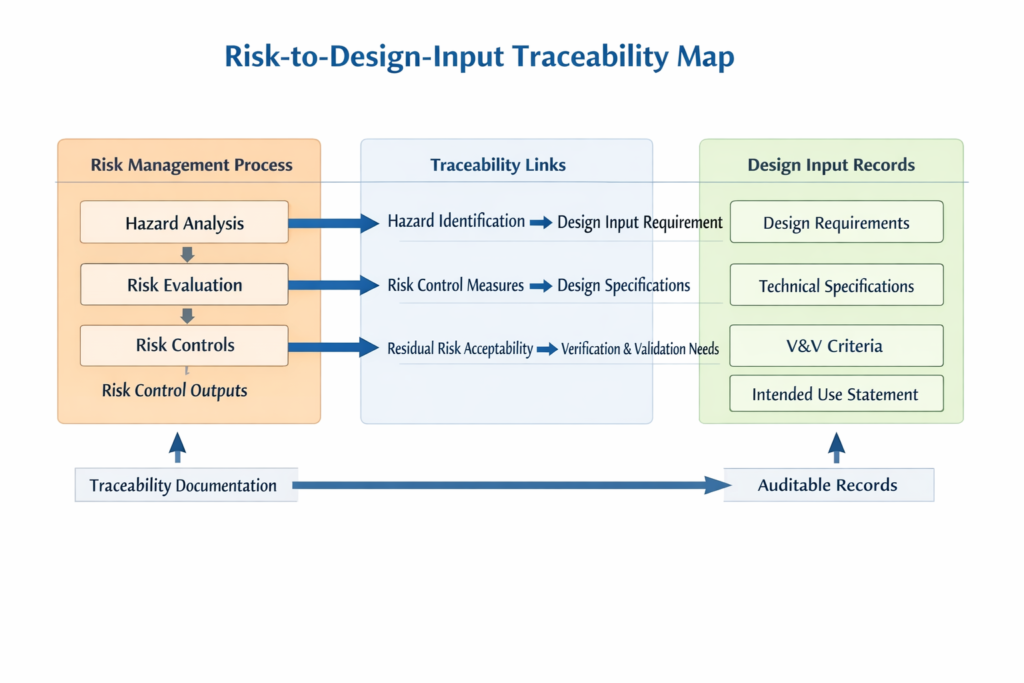
Outputs of risk management activities. This is the structural link to ISO 14971. Under legacy 820.30, ISO 14971 compliance was strongly recommended but was not codified as a design control deliverable. Clause 7.3.3 makes it mandatory: risk controls identified in the ISO 14971 risk assessment must appear as documented design inputs. The risk management process must be initiated before or concurrent with design input definition, and the traceability from risk file to design input record must be explicit and auditable.
Applicable regulatory and standard requirements. For dual-market manufacturers, this means the design input record must cite both FDA regulatory requirements and EU MDR General Safety and Performance Requirements where applicable.
A design input record carried over from 820.30(c) that lists performance specifications without ISO 14971 output citations and usability references does not conform to Clause 7.3.3 — regardless of how accurately the header now reads “7.3.3.”
There is a second problem at 7.3.3 that published industry analysis has identified but FDA has not formally addressed. Clause 7.3.3 absorbs a fragment of legacy 820.30(g): because intended use must be traced from design inputs forward, and validation under Clause 7.3.7 confirms the device meets intended use, the intended-use statement must be explicitly captured in the design input record. Manufacturers who mapped 820.30(g) only to Clause 7.3.7 without ensuring intended-use traceability begins at Clause 7.3.3 have an incomplete traceability chain. Clause reference reflects mapped standard requirement based on published practitioner analysis (Moon/Mathew, Drug Delivery Leader, 2024). Verify against current edition and FDA guidance before audit use.
Clause 7.3.6: Verification Plans Need Statistical Technique Documentation
Legacy 820.30(f) verification plans typically documented test methods and acceptance criteria. Many treated statistical technique selection as informal engineering judgment — sample sizes chosen by convention, confidence levels assumed rather than stated.
Clause 7.3.6 requires the verification plan to document all three prospectively: methods, acceptance criteria, and statistical techniques with rationale. The statistical technique documentation is not optional annotation. It is a plan deliverable that must exist before test execution begins. A verification plan that states “n=30, pass/fail per specification” without documenting why n=30 was selected, what confidence level it targets, and what reliability claim it supports does not satisfy the clause.
For devices that connect to or interface with other devices or systems, Clause 7.3.6 also requires verification to confirm performance when connected. Legacy 820.30(f) carried no explicit interface verification requirement. Most legacy DHFs for networked or connected devices lack this evidence entirely.
Clause 7.3.7: Validation Requires Representative Product Rationale — On Paper
Legacy 820.30(g) required validation on production or production-equivalent devices. Clause 7.3.7 requires validation on “representative product” — and requires the rationale for the choice of representative product to be recorded.
The recorded rationale is itself an auditable deliverable. It must document the basis for determining that the tested units represent the intended production output: lot selection criteria, manufacturing process equivalence evidence, worst-case analysis basis. Many legacy validation reports identified tested units as “representative” or “production-equivalent” without this documented rationale. Under ISO 13485:2016, the absence of the rationale document means the validation record is incomplete — even if the underlying testing was technically sound.
Clause 7.3.7 also requires clinical evaluations or performance evaluations “in accordance with applicable regulatory requirements.” For dual-market manufacturers, this means Clause 7.3.7 validation records must include clinical evidence satisfying EU MDR Annex XIV depth where CE marking is required — not merely 510(k) predicate-based equivalence data.

The Crosswalk Gap FDA Has Not Addressed
FDA’s CDRH published a QMSR Design and Development presentation describing requirements per Clause 7.3 sub-clause. The agency’s QMSR FAQ addresses general transition questions. But neither document provides an exhaustive clause-level crosswalk mapping every legacy 820.30(a)–(j) sub-element to specific ISO 13485:2016 Clause 7.3 sub-clause requirements with explicit gap identification.
The last formal FDA design control guidance documents — the 1997 Design Control Guidance for Medical Device Manufacturers and the 1999 Guide to Inspection of Quality Systems — have not been updated to reference QMSR or ISO 13485 clause structure as of March 2026. Industry practitioners note the 1997 waterfall model remains “fully compatible” with ISO 13485:2016, but FDA has not published a formal post-QMSR confirmation. Manufacturers relying on the 1997 guidance as their interpretive authority for QMSR compliance carry documentation risk in areas where Clause 7.3 requirements exceed legacy 820.30 scope.
The most consequential gap in the informal crosswalk is at Clause 7.3.3. Published analysis from named regulatory professionals identifies that Clause 7.3.3 absorbs requirements from both legacy 820.30(c) and a fragment of 820.30(g), creating a traceability obligation that spans two legacy regulation sections. No FDA document explicitly addresses this overlap.
Risk Management Is Now a Federal Regulatory Requirement
Before QMSR, FDA did not enforce ISO 14971 through 820.30. Risk management and design controls were parallel obligations — related in practice, separate in regulatory structure.
QMSR collapses that separation.
ISO 13485:2016 Clause 7.1 requires documented risk management processes throughout product realization, with the clause note referencing ISO 14971. Clause 7.3.3 requires risk management outputs as design inputs. Clause 7.3.7 validation must confirm the device meets intended use, which intersects with ISO 14971’s requirement for a residual risk acceptability determination.
The practical consequence: non-conformance with ISO 14971 process requirements — no documented risk management outputs, no risk controls traceable to design inputs, no residual risk determination cross-referenced in validation — is now also non-conformance with 21 CFR 820.10(a). An FDA 483 observation can cite the ISO 13485:2016 clause requirement directly.
Dual-Market Evidence: Two Frameworks, One Test Report
For manufacturers selling into both the US and EU markets, the same underlying V&V test data must serve two different evidence frameworks.
FDA investigators inspect the Design and Development File (Clause 7.3.10) and expect V&V records structured against ISO 13485:2016 sub-clauses. EU notified bodies assess technical documentation under MDR Annex IX and expect V&V records explicitly linked to Annex I GSPR compliance. The FDA framework traces V&V results back to design inputs (Clause 7.3.3). The EU framework traces V&V results forward to GSPR clause conformity.
In theory, this is solvable. A single V&V record set can serve both — if it explicitly cross-references both ISO 13485:2016 sub-clauses and EU MDR GSPR entries. Most legacy DHF records reference neither. No published T1 guidance from FDA, the European Commission, or IMDRF explicitly confirms whether a single DDF record satisfying both referencing conventions meets both regulatory frameworks simultaneously. Until that guidance appears, dual-market manufacturers building a unified DDF structure are working from reasonable inference, not confirmed regulatory position.
EU MDR Annex IX Chapter II requires notified body technical documentation assessment for Class III and Class IIb devices. Draft amendments under consultation in early 2026 propose limiting Class IIb TD assessment to one representative device per generic device group — potentially narrowing the audit scope but not eliminating the substantive V&V evidence requirement.
SaMD and IEC 62304: Where Software Meets Clause 7.3
For Software as a Medical Device, IEC 62304 provides the software lifecycle framework that maps directly onto Clause 7.3. The IEC 62304 Software Development Plan constitutes the software component of the Clause 7.3.2 Design and Development Plan. The Software Requirements Specification constitutes design inputs under Clause 7.3.3 — including risk-derived software requirements from IEC 62304 Clause 7.
IEC 62304 verification activities (unit, integration, and system-level testing) fulfill Clause 7.3.6, and IEC 62304 system testing fulfills Clause 7.3.7 for software functionality. The built-in traceability from requirements through test cases that IEC 62304 demands satisfies Clause 7.3.6 evidence requirements for software components.
The “representative product” question is harder for software. Clause 7.3.7 requires documented rationale for representative product selection. For SaMD, the validated software version, build configuration, and deployment environment (including cloud infrastructure and OS version where applicable) constitute the “representative product.” The rationale must document that the tested configuration represents the intended release configuration. FDA has not published specific guidance defining representative product rationale requirements for software-only devices — this remains an open implementation gap where manufacturers must construct defensible rationale without regulatory precedent. This characterisation is based on available IEC 62304 guidance and practitioner analysis. No T1 FDA guidance confirms representative product rationale requirements for SaMD specifically.
For AI/ML-enabled SaMD, FDA’s pre-QMSR framework expected Software Pre-Specifications and Algorithm Change Protocols mapping to Clause 7.3.9 (design changes). No post-QMSR update has appeared as of March 2026.
Closing the ISO 13485 Design Control Gaps: Implementation Sequence
Phase 1 — Clause-Level Gap Analysis
Month 1. Map every existing DHF section to Clause 7.3.1–7.3.10 by substantive content, not by header rename. For each active product, inventory V&V plans and flag any missing statistical technique documentation (7.3.6), representative product rationale (7.3.7), or risk management output citations in design inputs (7.3.3). The FDA CDRH QMSR Design and Development presentation (Slides 14–32) serves as the authoritative checklist.
Phase 2 — Procedure and Template Revision
Weeks 3–6. Revise the Design and Development Procedure to reference Clause 7.3 structure. The DDF template needs a sub-clause index covering 7.3.1–7.3.10 records. Verification and validation plan templates need pre-defined acceptance criteria with rationale, statistical technique documentation, and representative product rationale as mandatory fields — not optional appendices.
Phase 3 — ISO 14971 Integration
Months 2–3. Each active product’s risk file must explicitly identify risk controls that are design inputs. The traceability from risk file output to Clause 7.3.3 design input record must be documented and auditable. Without this link, the structural gap between risk management and design controls that QMSR made enforceable remains open.
Phase 4 — Record Remediation and Internal Audit
Months 2–4. Not every legacy record needs to be rewritten. Focus on products with active market authorisation. Where legacy validation was technically sound but documentation is incomplete — missing representative product rationale, for instance — create supplementary records documenting the rationale with contemporaneous evidence. Where legacy records cannot be remediated because acceptance criteria were never defined prospectively, document the gap, assess risk impact, and determine whether re-verification or re-validation is necessary.
By Month 4, conduct an internal audit of design control records against ISO 13485:2016 Clause 7.3 sub-clause requirements — not against legacy 820.30. The audit scope must include the four gap areas: Clause 7.3.3 inputs expansion, Clause 7.3.6 statistical technique documentation, Clause 7.3.7 representative product rationale, and ISO 14971 traceability integration.
What Remains Unresolved
Four specific gaps remain open:
The 820.30(c)-to-7.3.3 crosswalk anomaly — where Clause 7.3.3 absorbs obligations from both legacy 820.30(c) and 820.30(g) — has no FDA guidance addressing it. Manufacturers must build their own traceability logic and defend it at inspection.
Dual-market DDF architecture — whether a single record set cross-referencing both ISO 13485:2016 sub-clauses and EU MDR GSPR entries satisfies both FDA and notified body requirements simultaneously — remains unconfirmed by any T1 regulatory source.
Representative product rationale for SaMD has no FDA-published definition specific to software-only devices. Manufacturers must construct defensible rationale from IEC 62304 principles without regulatory precedent.
FDA’s 1997 and 1999 design control guidance documents remain the last published interpretive guidance for design controls. Neither has been updated for QMSR. Their continued applicability is assumed by practitioners but unconfirmed by the agency.
These are not speculative concerns. They are the specific points where an FDA investigator’s expectations and a manufacturer’s records are most likely to diverge — and where the absence of formal guidance transfers the compliance burden entirely to the manufacturer’s documentation.
Clause mapping reflects common audit practice and published regulatory analysis. Verify with your certification body and applicable regulatory authority for specific expectations.
This content is for educational purposes only and does not constitute certification, legal, or regulatory advice. Organizations should consult qualified professionals for implementation specific to their context.
About AEC International
AEC International provides ISO certification, training, and consultancy services at the intersection of medical device quality management, regulatory compliance, and risk management. We support organisations across industries in achieving and maintaining ISO certification — from gap analysis and implementation through audit preparation and continual improvement.
Learn more: www.aec.llc