
Quick Answer: The FDA QMSR, effective 2 February 2026, incorporates ISO 13485:2016 by reference and replaces the legacy 820.30 framework. Any ISO 13485 Clause 7.3 exclusion that cites 820.30 class-based exemptions now references superseded regulation. Contract manufacturers must reassess exclusion justifications under the current QMSR framework regardless of whether manufacturing activities changed.
The FDA QMSR became effective on 2 February 2026. For contract manufacturers and component suppliers holding ISO 13485 certification, this created a problem most have not yet addressed: the regulatory-permission basis underpinning their ISO 13485 Clause 7.3 exclusion no longer exists in its original form.
Organisations that excluded Clause 7.3 by citing 21 CFR 820.30 Class I device exemptions are now citing superseded regulation. The QMSR incorporates ISO 13485:2016 by reference, replacing the legacy 820.30 framework with ISO 13485’s own condition-based exclusion mechanism. The exclusion question has reopened — regardless of whether the organisation’s manufacturing activities changed. Every quality manual that references 820.30 is now exposed.
What Does Clause 7.3.1 Actually Require?
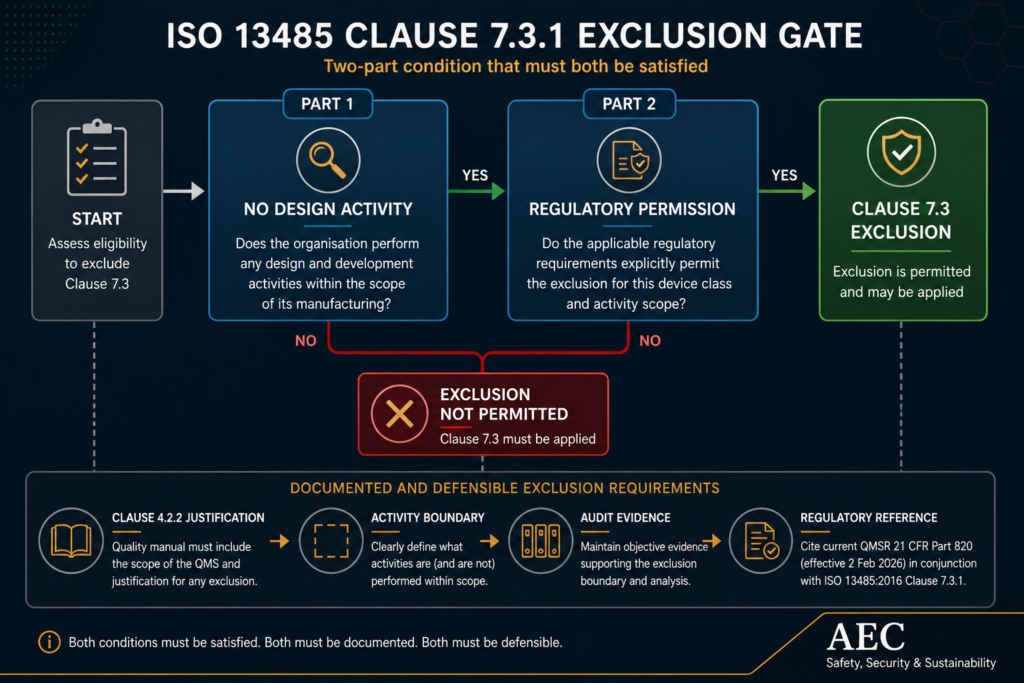
ISO 13485:2016 Clause 7.3.1 permits exclusion of design and development controls where “applicable regulatory requirements permit” it. This is not an activity test alone. It is a two-part gate.
The first test asks whether the organisation performs design activity. The second — and the one most quality manuals omit — asks whether the applicable regulatory framework explicitly permits the exclusion for that device class and activity scope. Both must be satisfied. Both must be documented.
Clause 4.2.2 requires the quality manual to include the scope of the QMS and “details of and justification for any exclusion.” An undocumented exclusion, or one justified only by a statement that “we manufacture to customer drawings,” fails this requirement on its face. The justification must name the regulatory framework, identify the device classification or scope limitation, and articulate the activity boundary.
Under the QMSR, the regulatory-permission reference must cite 21 CFR Part 820 as revised — not legacy 820.30. Quality manuals that still reference 820.30 class-based exemptions are citing a provision that the FDA’s December 2025 technical amendments formally replaced across multiple Class I exemption classifications.
Clause reference reflects mapped standard requirement. Verify against current edition before audit use.
Where Do Organisations Fail on Clause 7.3 Exclusions?
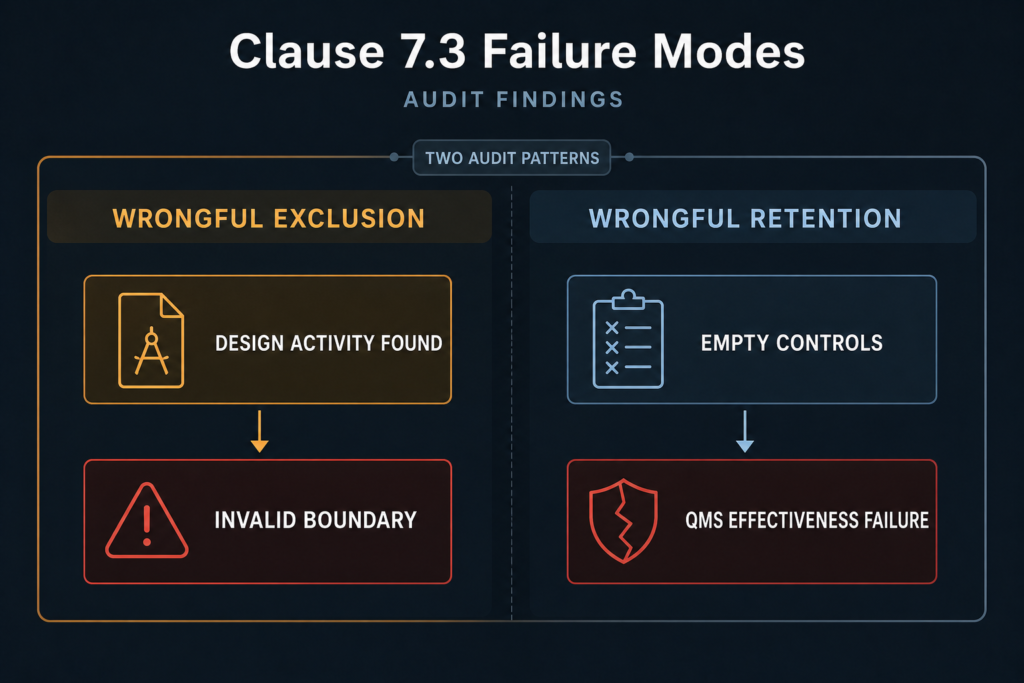
Auditors encounter two recurring failure modes on Clause 7.3 exclusions, and the QMSR transition has made both more exposed.
Wrongful exclusion is the more common finding. The organisation states it does not design the product — the customer provides the design. The quality manual contains no analysis of whether applicable regulatory requirements permit this exclusion, no reference to Clause 7.3.1 conditions, and no articulation of the boundary between receiving customer drawings and performing specification or customisation activities.
The auditor requests evidence of the design transfer process and discovers the organisation selected critical raw materials within performance tolerances, defined process parameters affecting device dimensional output, or adapted assembly sequences not specified in customer documentation. A major nonconformity follows: the exclusion is invalid because design activity occurred within the organisation’s scope.
Wrongful retention is less frequently cited but equally problematic. The organisation retains Clause 7.3 controls as a formality — empty design review forms with no substantive gate outcomes, a design history file with no linked risk management outputs, references to 7.3 controls for a device where no design activity occurs at that facility. Auditors reject these as procedurally compliant but substantively non-implemented. Keeping Clause 7.3 without actually running design controls is not a conservative choice. Clause 4.1 treats this as a QMS effectiveness failure.
The QMSR amplifies both patterns. FDA investigators operating under Compliance Program 7382.850 (effective 2 February 2026) will evaluate design activity claims against ISO 13485:2016 Clause 7.3 — not legacy QSIT procedures. Firms retaining 820.30 exclusion language without QMSR reassessment will be audited under the current framework regardless of prior FDA inspection history.
Where Does Design Activity Begin Under QMSR?
Here is the coverage gap that makes this topic genuinely difficult. No published FDA guidance document, QMSR preamble section, or FAQ response provides explicit criteria for determining when a contract manufacturer’s receipt of customer design inputs constitutes design activity triggering Clause 7.3.1 obligations. The FDA’s January 2026 Town Hall confirmed that contract manufacturers meeting the manufacturer definition cannot treat Clause 7.1 risk management obligations as inapplicable. But the ISO 13485 Clause 7.3 exclusion activity boundary was not defined.
The QMSR FAQ’s activity-scoping principle is directionally relevant: importers and distributors implement only QMS elements corresponding to their actual activities. Applied to contract manufacturers, this creates an activity-by-activity analysis — not a blanket exclusion or blanket inclusion. But this principle was stated in the context of importers and distributors, not Clause 7.3 specifically. It does not resolve the boundary.
This gap is real. It is unresolved in published T1 and T2 sources as of May 2026. Organisations asserting an exclusion boundary must do so without a regulatory definition to cite — which means the quality manual justification and the audit evidence supporting it carry the full burden of defensibility.
For organisations dual-registered under QMSR and EU MDR/IVDR, the exclusion logic diverges further. The EU framework does not permit a legal manufacturer to exclude Clause 7.3 on the basis that design inputs originated from a customer. The legal manufacturer bears full design obligation through MDR Annex I General Safety and Performance Requirements. A Clause 7.3 exclusion that holds under QMSR activity-based scoping may fail entirely under EU notified body certification. The two frameworks cannot be treated as equivalent.
Practical Steps
- Pull your quality manual exclusion statement now. Identify the Clause 4.2.2 justification for any Clause 7.3 exclusion. If it references 820.30, Class I exemptions, or any pre-QMSR language, the regulatory-permission basis requires immediate update.
- Map every activity at your facility that touches product specification. Material selection within performance tolerances, process parameter setting that affects device output, assembly sequence adaptation, and process validation choices that influence device function — all of these are design activity under Clause 7.3.1, regardless of who originated the design.
- Rewrite the exclusion justification to cite current regulatory framework. The Clause 4.2.2 statement must reference QMSR 21 CFR Part 820 (effective 2 February 2026) in conjunction with ISO 13485:2016 Clause 7.3.1. Where dual registration applies (QMSR + MDR/IVDR), document each regulatory framework’s exclusion analysis separately.
- If any design activity exists, activate Clause 7.3 controls with documented scope. Partial retention — applying design controls to the specific activities performed — is more defensible than blanket exclusion with weak justification. Define the scope boundary explicitly: what is covered, what is not, and why.
- Reference AAMI TIR102:2019 for clause-level mapping. This is the FDA-referenced mapping document between legacy 21 CFR Part 820 and ISO 13485:2016. Quality managers should use it to verify which ISO 13485 clauses correspond to superseded 820.30 provisions.
Key Takeaway
The ISO 13485 Clause 7.3 exclusion is not a historical decision that stays settled. It is a regulatory-permission claim that must be re-evaluated whenever the applicable regulatory framework changes — and the QMSR changed it on 2 February 2026. Contract manufacturers and component suppliers that excluded Clause 7.3 under 820.30 logic are now operating with an exclusion that cites superseded regulation. Whether or not your activities changed, the permission basis did. Reassess the exclusion, rewrite the quality manual justification, and prepare audit evidence for the activity boundary. The next surveillance audit will evaluate it under the current framework.
If your organisation needs support with ISO 13485 gap analysis, design controls training, or preparing for the QMSR transition alongside your existing design controls framework, AEC International provides certification consultancy across the medical device sector.